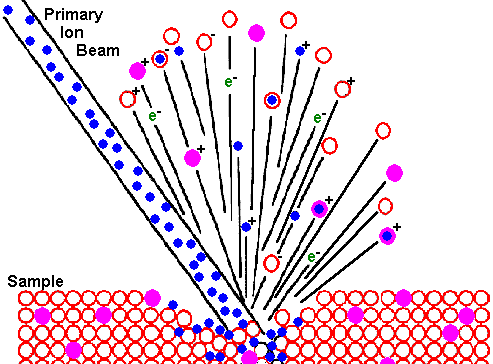
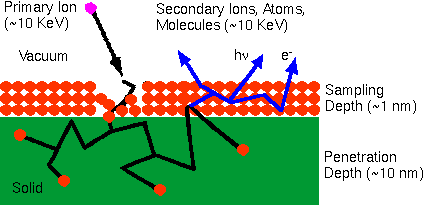
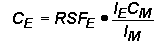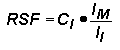Secondary Ion Mass Spectrometry Theory TutorialIntroductionBombardment of a sample surface with a primary ion beam followed by mass spectrometry of the emitted secondary ions constitutes secondary ion mass spectrometry (SIMS). The best SIMS reference is Secondary Ion Mass Spectrometry: Basic Concepts, Instrumental Aspects, Applications, and Trends, by A. Benninghoven, F. G. Rüdenauer, and H. W. Werner, Wiley, New York, 1987 (1227 pages).
Uses for SIMSToday, SIMS is widely used for analysis of trace elements in solid materials, especially semiconductors and thin films. The SIMS ion source is one of only a few to produce ions from solid samples without prior vaporization. The SIMS primary ion beam can be focused to less than 1 um in diameter. Controlling where the primary ion beam strikes the sample surface provides for microanalysis, the measurement of the lateral distribution of elements on a microscopic scale. During SIMS analysis, the sample surface is slowly sputtered away. Continuous analysis while sputtering produces information as a function of depth, called a depth profile. When the sputtering rate is extremely slow, the entire analysis can be performed while consuming less than a tenth of an atomic monolayer. This slow sputtering mode is called static SIMS in contrast to dynamic SIMS used for depth profiles. Shallow sputtering minimizes the damage done to organic substances present on the sample surface. The resulting ion fragmentation patterns contain information useful for identifying molecular species. Only dynamic SIMS will be treated in this surface analysis computer aided instruction package because only dynamic SIMS yields quantitative information.
Ion Beam SputteringThe bombarding primary ion beam produces monatomic and polyatomic particles of sample material and resputtered primary ions, along with electrons and photons. The secondary particles carry negative, positive, and neutral charges and they have kinetic energies that range from zero to several hundred eV.
Primary beam species useful in SIMS include Cs+, O2+, O , Ar+, and Ga+ at energies between 1 and 30 keV. Primary ions are implanted and mix with sample atoms to depths of 1 to 10 nm. Sputter rates in typical SIMS experiments vary between 0.5 and 5 nm/s. Sputter rates depend on primary beam intensity, sample material, and crystal orientation. The sputter yield is the ratio of the number of atoms sputtered to the number of impinging primary ions. Typical SIMS sputter yields fall in a range from 5 and 15.
Sputtering EffectsThe collision cascade model has the best success at quantitatively explaining how the primary beam interacts with the sample atoms. In this model, a fast primary ion passes energy to target atoms in a series of binary collisions. Energetic target atoms (called recoil atoms) collide with more target atoms. Target atoms that recoil back through the sample surface constitute sputtered material. Atoms from the sample's outer monolayer can be driven in about 10 nm, thus producing surface mixing. The term knock-on also applies to surface mixing.
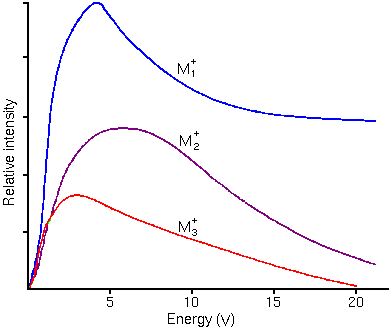
Sputtering leads to surface roughness in the sputter craters. Lattice imperfections, either already present or introduced by surface mixing, can be germs for roughness that takes the form of ribbons, furrows, ridges, cones, and agglomerations of cones. Polycrystalline materials form rough crater bottoms because of differential sputter rates that depend on crystal orientation. Secondary Ion Energy DistributionsThe sputtering process produces secondary ions with a range of (translational) kinetic energies. The energy distributions are distinctly different for atomic and molecular ions. Molecular ions have relatively narrow translational energy distributions because they have kinetic energy in internal vibrational and rotational modes whereas atomic ions have all kinetic energy in translational modes. The following figure shows typical energy distributions for mono, di, and triatomic ions.
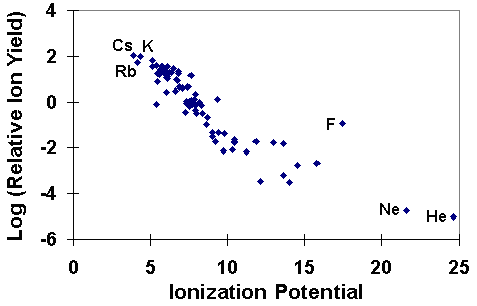
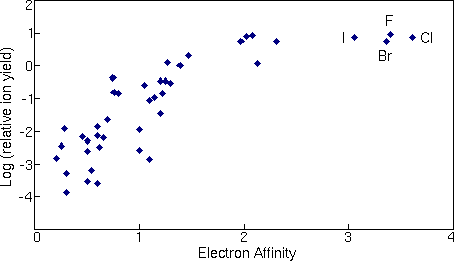
Secondary Ion Yields -- Elemental EffectsThe SIMS ionization efficiency is called ion yield, defined as the fraction of sputtered atoms that become ionized. Ion yields vary over many orders of magnitude for the various elements. The most obvious influences on ion yield are ionization potential for positive ions and electron affinity for negative ions. For example, the following figure shows the logarithm of positive ion yields plotted as a function of ionization potential. The ion yields are relative to silicon in a silicon matrix with oxygen sputtering.
The correlations of ionization potential with secondary ion yields are not perfect. Variations depend both on the sample matrix and on the element itself. For example, the presence of oxygen in the sample enhances positive ion yields for most elements, but fluorine exhibits anomalously high positive ion yields in nearly all samples. Some elements, such as helium and neon fall outside the trend shown in the picture. The next figure shows a similar treatment for negative ions where the logarithms of relative ion yields are plotted against electron affinities. The ion yields are relative to silicon for measurements in a silicon matrix with cesium ion sputtering. The four halides are the elements that deviate furthest from the trend line.
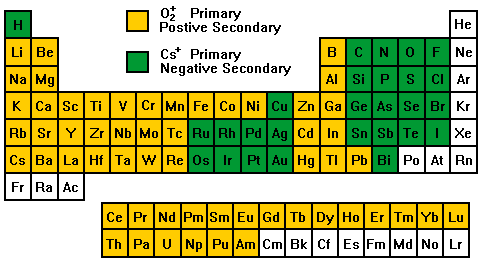
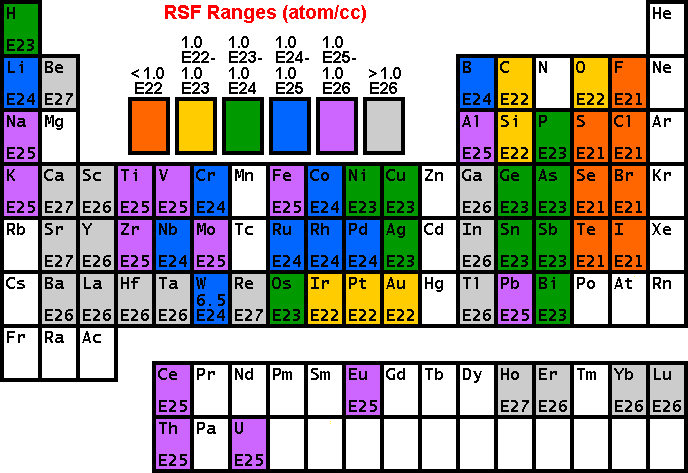
Secondary Ion Yields -- Primary Beam EffectsOther factors affect the secondary ionization efficiencies in SIMS measurements. Oxygen bombardment increases the yield of positive ions and cesium bombardment increases the yield of negative ions. The increases can range up to four orders of magnitude. Oxygen enhancement occurs as a result of metal-oxygen bonds in an oxygen rich zone. When these bonds break in the ion emission process, the oxygen becomes negatively charged because its high electron affinity favors electron capture and its high ionization potential inhibits positive charging. The metal is left with the positive charge. Oxygen beam sputtering increases the concentration of oxygen in the surface layer. The enhanced negative ion yields produced with cesium bombardment can be explained by work functions that are reduced by implantation of cesium into the sample surface. More secondary electrons are excited over the surface potential barrier. Increased availability of electrons leads to increased negative ion formation. The variability in ionization efficiencies leads to different analysis conditions for different elements as indicated on the periodic table.
Relative Sensitivity FactorsQuantitative analysis by SIMS uses relative sensitivity factors defined according to the following equation.
The major (or matrix) element is usually chosen as the reference. Substituting M (matrix) for R (reference) and rearranging gives the following equation.
In trace element analysis, we can assume that the matrix elemental concentration remains constant. The matrix concentration can be combined with the elemental RSF to give a more convenient constant, RSF.
This RSF is a function of the element of interest and the sample matrix.
Note that RSF and CE have the same concentration units. This is the most common form of the RSF equation.
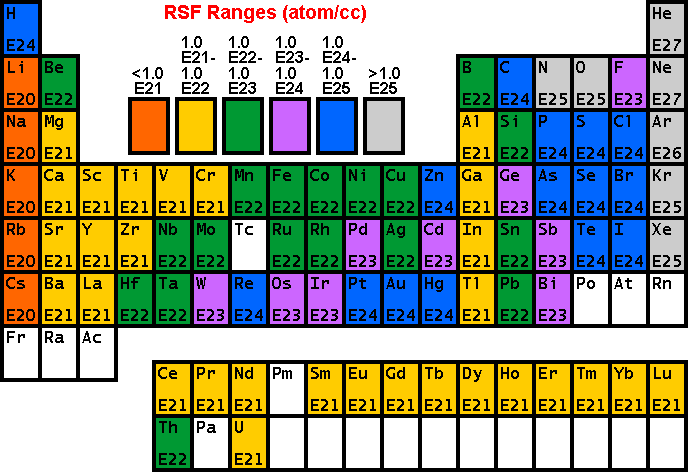
RSF TablesThe following tables of RSF values
(from R.G. Wilson, Int. J. Mass Spectrometry. Ion Proc., 143,
43, 1995) show how sensitivity depends on the element of interest. Low
RSFs mean high sensitivity. Note that modest concentrations of high
sensitivity elements can saturate electron multiplier ion detectors. The following RSFs have been measured for oxygen primary ion bombardment, positive secondary ions, and a silicon matrix.
These RSFs have been measured for cesium primary ion bombardment, negative secondary ions, and a silicon matrix.
Sensitivity and Detection LimitsThe SIMS detection limits for most trace elements are between 1e12 and 1e16 atoms/cc. In addition to ionization efficiencies (RSF's), two other factors can limit sensitivity. The output of an electron multiplier is called dark counts or dark current if no secondary ions are striking it. This dark current arises from stray ions and electrons in instrument vacuum systems, and from cosmic rays. Count rate limited sensitivity occurs when sputtering produces less secondary ion signal than the detector dark current. If the SIMS instrument introduces the analyte element, then the introduced level constitutes background limited sensitivity. Oxygen, present as residual gas in vacuum systems, is an example of an element with background limited sensitivity. Analyte atoms sputtered from mass spectrometer parts back onto the sample by secondary ions constitute another source of background. Mass interferences also cause background limited sensitivity.
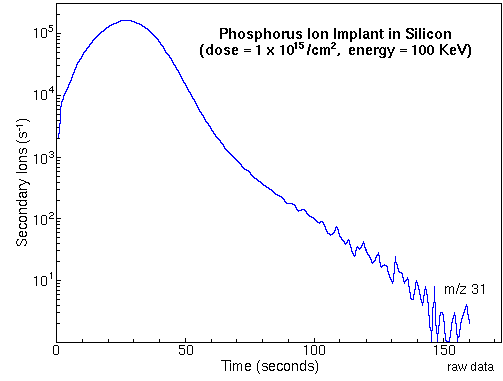
Depth ProfilingMonitoring the secondary ion count rate of selected elements as a function of time leads to depth profiles. The following figure shows the raw data for a measurement of phosphorous in a silicon matrix. The sample was prepared by ion implantation of phosphorous into a silicon wafer. The analysis uses Cs+ primary ions and negative secondary ions.
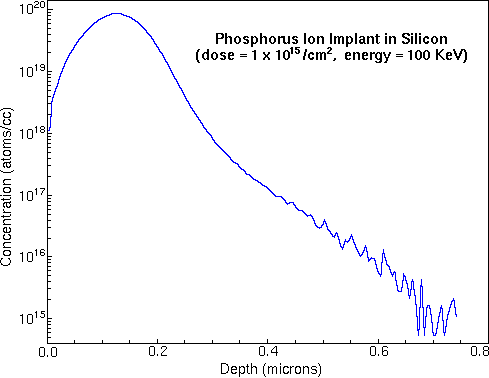
To convert the time axis into depth, the SIMS analyst uses a profilometer to measure the sputter crater depth. A profilometer is a separate instrument that determines depth by dragging a stylus across the crater and noting vertical deflections. At the end of the above phosphorous depth profile, profilometry gives 0.74 um for the crater depth. Total crater depth divided by total sputter time provides the average sputter rate. Relative sensitivity factors (RSFs) convert the vertical axis from ion counts into concentration. The appropriate RSF value for the above phosphorous implant is 1.07E23 atoms per cubic centimeter and the matrix current (IM) is 2.2E8 silicon ion counts per second. The following figure shows the above phosphorous depth profile plotted on depth and concentration axes.
Depth ResolutionDepth resolution depends on flat bottom craters. Modern instruments provide uniform sputter currents by sweeping a finely focused primary beam in a raster pattern over a square area. In some instruments, apertures select secondary ions from the crater bottoms, but not the edges. Alternatively, the data processing system ignores all secondary ions produced when the primary sputter beam is at the ends of its raster pattern.
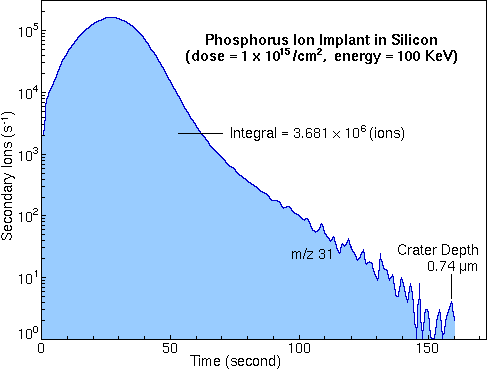
Standards for RSF MeasurementQuantitative SIMS analysis requires standard materials from which to measure RSF values. Because ion yields depend on the analyte element, the sputtering species, and the sample matrix, separate RSF's must be measured for each. Ion implants are good standards. It is possible to implant virtually any element into any matrix. Ions can be passed through a mass analyzer before implantation to insure implant purity. Typical implant ion energies range from 50 to 300 keV. Higher energies are usually used for heavier ions, producing typical implant depths centered around 0.2 um. Most importantly, the implant ion current can be integrated to determine total ion dose. However, care must be taken to exclude secondary electron and ion currents from the total measurement. The phosphorous implant raw data shown in the Depth Profile section can be used to calculate an RSF value for phosphorous in silicon. The shaded area is the total signal from the phosphorous implant (3.68e6 phosphorous ions).
Dividing the total signal by the measurement time gives Ii, the average phosphorous secondary ion signal during the measurement (2.69e4 / s). The implant dose (1e15 ions per square centimeter in this example) and the crater depth (0.74 um) are required to calculate the average implant concentration, CI (1.35e19 atom/cc).
Inclusion of the silicon matrix current, IM, (2.18E8 / s) provides for RSF calculation according to the rearranged RSF equation.
The calculated RSF value (1.09e23) is in close agreement with the value cited (1.07e23) in the RSF Tables section.
Bulk AnalysisFor samples with homogeneously dispersed analyte, bulk analysis provides better detection limits than depth profiling, usually by more than an order of magnitude. Faster sputter rates increase the secondary ion signal in bulk analysis. The fastest possible sputtering requires intense primary ion beams which sacrifice depth resolution because they cannot be focused as required for flat bottom (rastered) craters. Otherwise, bulk analyses are similar to depth profiles. Ion intensity data are displayed as a function of time. This provides a means for verifying that the sample is indeed homogenous. In a typical heterogeneous sample, the analyte is concentrated in small inclusions that produce spikes in the data stream.
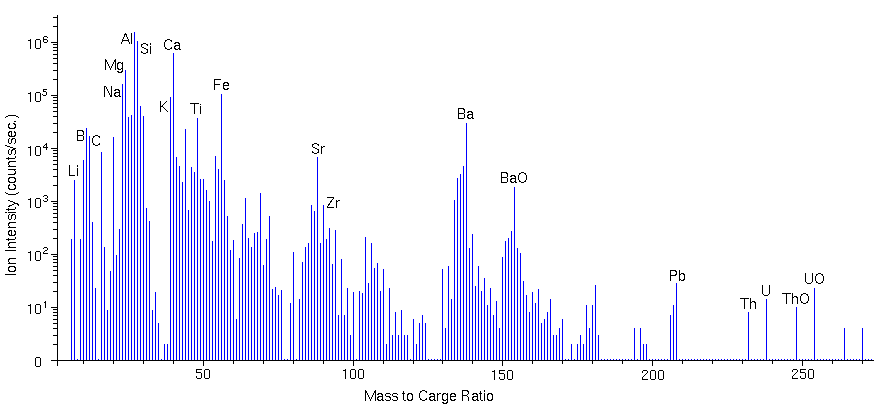
Mass SpectraMass spectra sample the secondary ions in a preselected mass range by continuously monitoring the ion signal while scanning a range of mass-to-charge (m/z) ratios. The mass analyzer can be either a magnetic sector or a quadrupole. The mass spectrum detects both atomic and molecular ions. Secondary ions containing more than one atom are called molecular ions in SIMS. Note that the term molecular ion also finds use in organic mass spectrometry where it refers to the parent ion before any fragmentation. The mass analyzer must be scanned in small steps to insure that all mass-to-charge (m/z) ratios are sampled. Ten steps per mass unit are common. At higher mass resolution, ten mass increments per peak width adequately define the peak shape. A mass spectrum with a mass range of 100 has at least 1000 data channels for which a reasonable analysis time is 0.1 s per channel. The following figure shows a compressed mass spectrum for a coal fly ash particle. The spectrum clearly shows Li, Be, B, C, O, Na, Mg, Al, Si, K, Ca, Ti, Fe, Zr, Ba, Pb, Th, U. Notable molecular ions include TiO, FeO, BaO, ThO, and UO at m/z 64, 72, 154, 248, and 254, respectively. The ion intensities reflect the isotopic abundances of the elements. For example, silicon isotopes intensities at m/z 28, 29, and 30 parallel the relative silicon natural abundances 92.2 : 4.7 : 3.1. Note the peak for doubly charged Ca++ at m/z 20.
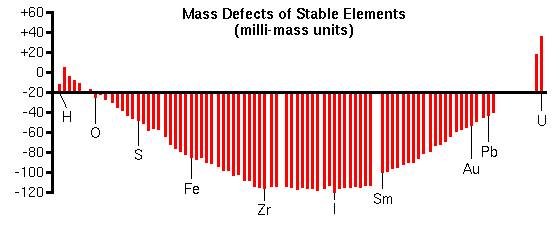
Mass InterferencesMass interferences occur whenever another ion has the same nominal mass as the analyte ion. Such interferences are called isobaric. During the analysis of iron in silicon for example, (28Si2)+ interferes because a singly-ionized molecule with two 28Si atoms has the same mass as 56Fe+ (m/z is 56 in both cases). Oxides are common interferences since oxygen-metal bonds are particularly stable. Thus, 40CaO+ can also interfere with 56Fe+ measurements. Low intensity hydrides of many elements appear one mass unit higher than the elements themselves. A good example is silicon-30 hydride (30SiH+) which interferes with trace phosphorous analysis. Primary ions often combine with sample elements to produce interferences. For example 133Cs32S2- is isobaric with Au- (m/z 197) during the measurement of gold in pyrite (FeS2). High Mass ResolutionAlthough analyte/interference pairs have the same nominal masses, their exact masses differ by a fraction of a mass unit. The exact mass minus the nominal masses is called the mass defect. Mass defects arise from differences in the nuclear binding energies that hold the protons and neutrons together in the nucleus. Mass defects vary from +0.0078 for hydrogen to 0.1 for elements in the middle of the periodic table to +0.051 for uranium.
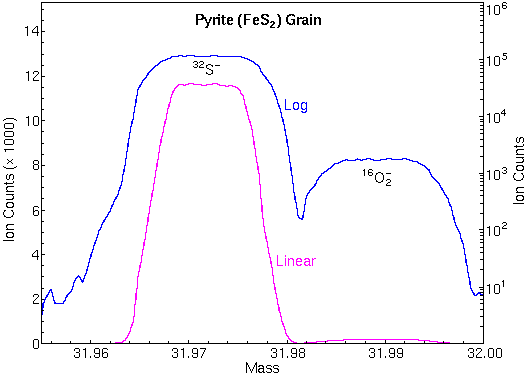
The curve of mass defects gives atomic ions higher masses than molecular interferences at relatively low masses and the opposite at higher masses. The following mass spectrum in the m/z 32 region shows separation of 32S and 16O2
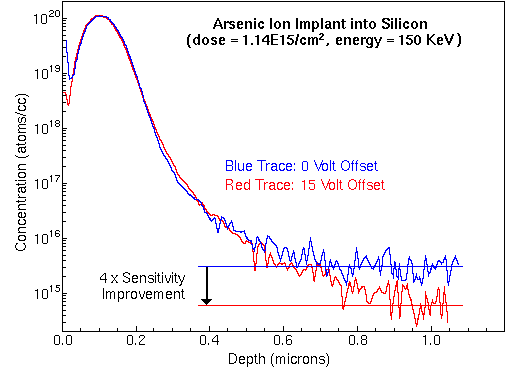
Mass spectrometers with sufficient mass resolution can separate atomic ions from molecular ion interferences. Mass resolution is usually specified in terms of m/delta m where m is the nominal mass of the two ions and delta m is their difference. For example, 56Fe and 28Si2 (m/z 55.9349 and 55.9539) require m/delta m 5,600 for separation while Au and 133Cs32S2 (m/z 196.9666 and 196.8496) require m/delta m 1700. These two isobaric pairs illustrate the tendency for atomic ions to have lower masses than molecular interferences at relatively low masses and the opposite at higher masses. Each different kind of SIMS mass analyzers has a range of possible mass resolutions. For example, a (well tuned) double focusing magnetic sector instrument can have mass resolution in the range m/delta m 500 to 10,000. Loss of secondary ion intensity accompanies operation at the high end of the mass resolution range. Minor IsotopesMinor isotopes can often resolve interference problems. However, minor isotope secondary ion intensities are lower (by the isotope ratio), detection limits are correspondingly higher (worse), and the RSF values must be adjusted (up). For example, measurement of iron using the 54Fe isotope avoids the interference of 28Si2 with 56Fe, but the 54Fe signal intensity is lower than 56Fe by the 54Fe/56Fe isotope ratio (0.0645). Natural abundance isotope ratios cannot always be used for these adjustments because some processes, such as ion implantation of a specific isotope, alter the ratios. Elemental InterferencesIn a few cases, an isotope of one element has the same nominal mass as an isotope of another. Their separation requires ultra-high mass resolution, beyond the capability of any commercial SIMS instrument. For example, 104Ru and 104Pd would interfere with each other and require m/delta m ~75,000 to separate. Fortunately, non-interfering isotopes are available for most elemental interferences. Voltage OffsetSputtering produces secondary ions with a distribution of energies. The voltage offset technique uses the energy analyzer of a mass spectrometer to select secondary ions from the high range of translational energies. The offset is simply a reduction in acceleration voltage. The energy analyzer deflects lower energy ions by a larger angle. A physical barrier (the inner jaw of the energy slit) intercepts the low energy ions. Ions that started with higher translational energies pass through to the mass analyzer. Voltage offset thus discriminates against molecular interferences relative to atomic species. The operator selects the energy cutoff point. A 30 V offset would eliminate essentially all of the di and triatomic ions from a typical distribution of energies. There is an analytical trade-off for this reduction of isobaric interferences. Most of the monatomic ions are also eliminated. A 30 V offset typically reduces the monatomic ion intensity by a factor of ten. The analyst must decide whether this reduction of the analyte signal is tolerable. Reduction of analyte signal intensity can usually be compensated by changing other experimental parameters, higher primary beam current or wider spectrometer slits, for example. The following figure shows the analysis of arsenic in silicon, both with and without voltage offset. A small interfering signal from 28Si30SiOH falls at m/z 75. Note the improved detection limits with 15 volt offset.
Sample ChargingThe SIMS primary ion beam, secondary ions, and secondary electrons produce a net electric current at the sample surface. If the sample material conducts, the current flows through the sample into the instrument. However, insulating samples undergo charge buildup. Sample charging diffuses the primary beam and diverts it from the analytical area, often eliminating the secondary ion signal entirely. Sample charging also changes the energy distribution of the secondary ions, which effects their transmission and detection by the mass spectrometer. When the sample is a thin dielectric on a conducting substrate, a strong electric field develops. Mobile ions such as sodium and lithium migrate in the electric field and depth profiles no longer reflect the original compositions of the layers. Several techniques are available to manage sample charging, and they are often used in combination. Electron BombardmentElectrons compensate for positive charge buildup that results from positive primary ions and/or negative secondary ions and electrons. Low energy electron beams work better because higher energies produce more than one secondary electron for every incoming electron. Low energy electron beams are more easily implemented in quadrupole SIMS instruments, making quadrupoles the system of choice for insulating materials. In contrast to quadrupoles, magnetic sector instruments maintain the sample at high positive potential for positive ion spectroscopy, making it difficult to bring in a low energy electron beam. High energy electron beams, though less effective, are widely used. Adjacent ConductorsConducting grids placed over the sample reduce the effects of charging on ion optics and bring a source of electrons near to positively charged areas of the sample. When struck by a primary ion, the conductors emit secondary electrons that migrate to the charging area. Similarly, samples are often coated with conducting materials such as gold or carbon. Before starting the analysis, the coating must be sputtered away, but only in the analytical area. Negative Primary Ion BeamsThe most common negative primary ion beam is O, available from the same duoplasmatron sources that more commonly produce O2+. Primary O beams find wide use for insulating geological samples. Automatic Voltage OffsetA continuously variable voltage offset can be applied to the accelerating voltage for samples that are only slightly charging. Automatic voltage offset procedures (called autovolt) are often incorporated into instrument control software. After every cycle in a depth profile analysis, the software invokes an energy distribution measurement and adjusts the voltage offset as needed to keep the peak of the distribution constant.
Ion ImagingIon images show secondary ion intensities as a function of location on sample surfaces. Image dimensions vary from 500 um to less than 10 um. Ion images can be acquired in two operating modes, called ion microscope or stigmatic imaging, and ion microbeam imaging or raster scanning. Ion microscopy requires a combination ion microscope/mass spectrometer capable of transmitting a mass selected ion beam from the sample to the detector without loss of lateral position information. Image detectors indicate the position of the arriving ions. Ion microscope images are usually round because the ion detectors are round. Lateral resolutions of 1 um are possible. A SIMS analyst selects images with higher lateral resolution at the expense of signal intensity and higher mass resolution at the expense of image field diameter. For ion microbeam imaging, a finely focused primary ion beam sweeps the sample in a raster pattern and software saves secondary ion intensities as a function of beam position. Microbeam imaging uses standard electron multipliers and image shape follows raster pattern shape, usually square. Lateral resolution depends on microbeam diameter and extends down to 20 nm for liquid metal ion guns. Some instruments simultaneously produce high mass resolution and high lateral resolution. However, the SIMS analyst must trade high sensitivity for high lateral resolution because focusing the primary beam to smaller diameters also reduces beam intensity.
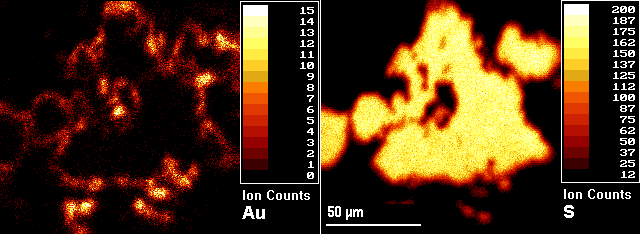
The example (microbeam) images show a pyrite (FeS2) grain from a sample of gold ore with gold located in the rims of the pyrite grains. The image on the right is 34S and the left is 197Au. The numerical scales and the associated colors represent different ranges of secondary ion intensities per pixel. Three-dimensional analyses are possible by acquiring images as a function of sputtering time (image depth profiles). Microscope sputtering rates exceed microbeam rates, often by several orders of magnitude. Thus microscope imaging produces depth scales more compatible with the scale of the lateral images. Microbeam imaging usually provides a better combination of image features, except when faster sputtering is required for three-dimensional analysis or for removing an overlayer before image acquisition.
Isotope Ratio MeasurementsIsotope ratio measurements are operationally similar to depth profiles except that precision and accuracy requirements are higher. Since all of the isotopes of an element have the same chemical properties, ionization and detection efficiencies remain nearly constant for the different isotopes. Precisions of 0.1% are commonplace and accuracies approach precisions. Error analysis indicates that the precisions are limited mainly by Poisson counting statistics. To attain these accuracies, SIMS instruments must be carefully tuned, and interferences must be eliminated. Mass spectral peaks should have flat tops and steep sides so that slight magnet instability does not change the ion signal intensity. The figure shows the necessary flat top peaks and the high mass resolution used for elimination of O2 interferences at m/z 32. Both were necessary for measuring accurate 34S/32S isotope ratios.
The ratio of isotope signal intensities must be corrected for slight variations in detection efficiency at different masses, and for slight variations that depend on signal intensity. These corrections are usually larger than the range of expected isotope ratios.
|